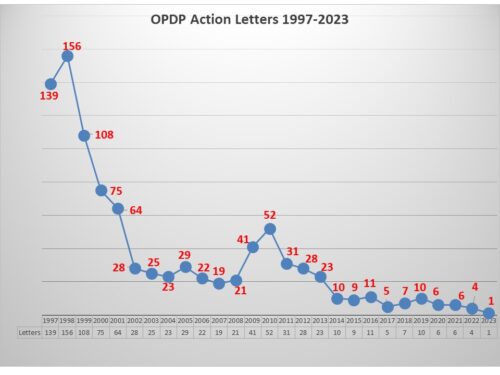
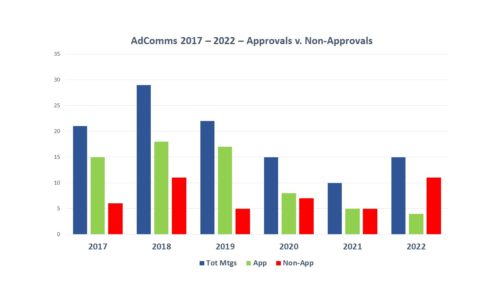
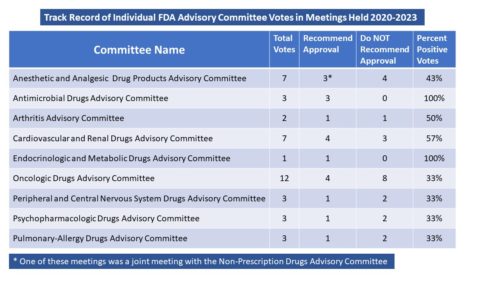
Every six months, we do a look-back to see what has changed in the way that FDA is communicating. One might not think there is much difference over time, but there is. For example, the air has pretty much gone out of the COVID balloon. And there is more to tell.
How many? First of all, consider volume. The number of press releases issued by FDA during the first six months of the year declined to 101 from the 125 issued by June 30 of last year, the lowest number at mid-year since 2017. In 2018, FDA issued a much larger number of releases – trend that continued through 2022, all years with much larger numbers than were seen in 2017.
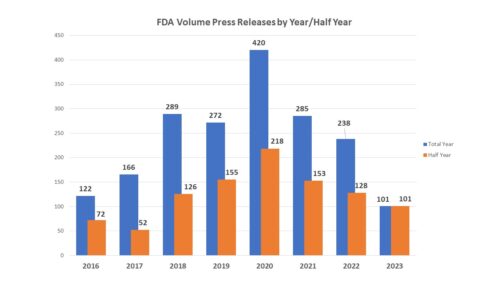
The increase can be attributable to two factors. First, the upward tick in releases beginning in 2017 that climbed steeply in 2018 and following slightly the following year matches the tenure of Dr. Gottlieb as FDA Commissioner. During his time at the post, the Commissioner’s office turned the relatively rare practice of issuing statements from the commissioner to a very common one. The second spike occurred in 2020, coinciding with the COVID-19 pandemic. In 2022, the agency issued 27 releases that involved the subject of COVID, 18 of which were during the first half of the year. In 2023, there were just 4.
One factor in the decrease is of course the near-absence of COVID-related news from the agency. But one cannot talk about volume without talking about the different way that FDA is putting out news – specifically the “FDA Roundup“. Prior to 2022, FDA posted news that did not quite meet the press release threshold through a mechanism called “FDA in Brief” which were halted in 2022 and seemingly replaced by the “FDA Roundup” which is housed on the press release page, but which contains a hodgepodge of news items covering actions by the agency during the past few days such as workshops being held or articles that have been published. While the FDA in Brief documents covered only one topic, the FDA Roundup can cover several and includes an update on COVID-related activities, meaning that while the overall volume of releases is down, the overall volume of information may, in fact, be up.
What did they talk about? Looking to press releases by category, there was
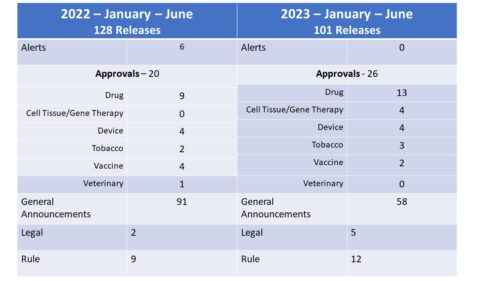
So far this year, the agency has had releases in the following categories with a comparison the first six months of last year. There were fewer releases this year, mostly due to a drop in the General Announcement category the bulk of which are the Daily Roundups. There was an increase in the number of approval related announcements (though it should be noted that in the case of Tobacco, the releases were largely in regard to the denial of approval). In 2023 so far, there have also been an increase in Legal (Warnings, e.g.) and Rule (guidance docs, proposed rules). Also of note, while there were 6 alerts in the first half of 2022, there have been none in 2023.
Did they speak Spanish? There are two large Spanish-language television networks operating in the United States, as well as weekly, semi-weekly and daily publications. Over the years of these look-backs, one thing that has stuck out about the press releases is that some of them are translated into Spanish, while many are not. Yet there seemed no obvious way to discern which would be bilingual and which would not. In the first half of 2023, there were 35 releases that were in both Spanish and English – about one-third of the releases overall. In the first half of 2022, there were 44 bilingual releases – also about one-third. But in 2020, there were only 34 bilingual releases out of the total of 154, or about one-fifth. (The Roundups are not bilingual.) It would appear that there may be an indication that FDA is upping the effort here to reach this important audience, though there is still a way to go. And the increase in profile of Spanish releases would also be higher when there are fewer press releases if the number of bilingual communications remained the same.
Overall, there have been shifts in the way the agency is communicating. We’ll be back at year end to see how we netted out.
Photo by AbsolutVision on Unsplash